

Parceria GIA-ATGC
No dia 09 de fevereiro de 2021 foi finalmente assinado o Termo de Compartilhamento estabelecido entre a UFPR, através de...

SEQUENCIAMENTO DE NOVA GERAÇÃO PARA ANÁLISES METAGENÔMICAS: ENFOQUE AO USO DO SEQUENCIADOR ILLUMINA
As novas tecnologias de sequenciamento, denominadas de tecnologias de sequenciamento de nova geração (Next Generation Sequencing-NGS), começaram a ser comercializadas em 2005 e estão evoluindo rapidamente. Todas essas tecnologias promovem o sequenciamento de DNA em plataformas capazes de gerar informação sobre milhões de pares de bases em uma única corrida.
Apesar de se diferenciarem consideravelmente entre si todos os sequenciadores de NGS se baseiam no processamento paralelo massivo de fragmentos de DNA. Enquanto, um sequenciador de eletroforese processa, no máximo, 96 fragmentos por vez, os sequenciadores de nova geração podem ler até bilhões de fragmentos ao mesmo tempo.
As plataformas de sequenciamento de nova geração são uma alternativa poderosa para estudos de genômica estrutural e funcional.
Atualmente, os sequenciadores estão divididos em 1ª geração (Degradação química e Interrupção da cadeia-Sanger), 2ª geração (454-Roche, Illumina Genome Analyser-HiSeq/MiSeq e SOLID), 3ª geração (Ion Torrent e PacBio RS) e 4ª geração (Nanopore).

Figura 1: Equipamentos utilizados para sequenciamento de DNA. 1ª geração (A) Sanger; 2ª geração (B) 454-Roche, (C) Illumina HiSeq, (D) Illumina MiSeq, (E) Solid; 3ª geração (F) Ion Torrent, (G) PacBio e 4ª geração (H) Nanopore.
Análises metagenômicas tradicionais utilizam a amplificação, clonagem e determinação da sequência nucleotídica de DNA ribossomal, pelo método de sequenciamento de DNA proposto por Sanger na década de 70. Para isso, é utilizada polimerização de DNA com incorporação de dideóxinucleotídeos marcados, método que foi grandemente automatizado na última década e meia, permitindo a execução dos diversos projetos-genoma. Porém, abordagens metagenômicas modernas aplicam distintas técnicas de sequenciamento de segunda geração, que permitem estudar genomas microbianos completos a partir de amostras ambientais, bem como identificar e pesquisar clusters gênicos funcionais codificando enzimas microbianas de interesse tecnológico, como lipases, despolimerases ou hidrolases para polímeros complexos.
O termo metagenômica é caracterizado pela aplicação de técnicas de biologia molecular que possibilitam a análise e caracterização de comunidades microbianas diretamente do ambiente natural, de forma independente do isolamento e cultivo dos microrganismos. Esse tipo de abordagem vem aprimorando e enriquecendo estudos filogenéticos sobre o potencial biotecnológico de microrganismos antes não acessados e/ou desconhecidos.
Embora distintas, as técnicas de Sanger e NGS apresentam vantagens e desvantagens, assim como limitações de custo/benefício para obter e gerar dados. A escolha do método depende principalmente do objetivo do trabalho proposto.
A principal vantagem do método de Sanger é, sem dúvida, o maior tamanho dos reads gerados e a precisão da base gerada (base calling), que tende a ser 10x maior que nos métodos NGS; já para os métodos de segunda geração, a principal vantagem consiste na construção in vitro de bibliotecas genômicas sem amplificação de fragmentos de DNA e sem clonagem, como por exemplo, em Escherichia coli. Portanto, em análises metagenômicas, é viável a utilização desses sequenciadores de segunda geração, pois permite a construção de grandes bibliotecas. Um dos principais sequenciadores indicado para este tipo de análise é o Illumina.
O sequenciador Illumina Genome Analyzer (Figura 2) produzido em 2006, baseia-se no conceito de “sequenciamento por síntese” o qual requer que as sequências a serem determinadas sejam convertidas numa biblioteca de sequenciamento especial, permitindo a amplificação e imobilização das sequências para serem submetidas à sequenciamento. Para este propósito, dois adaptadores diferentes são adicionados às terminações 5’ e 3’ de todas as moléculas.

Figura 2: Sequenciador Illumina MiSeq/HiSeq.
A biblioteca de cadeia dupla é desnaturada para obter DNAs de cadeia única. Estas cadeias simples são dispostas em concentrações muito baixas pelos canais de uma célula de fluxo. Esta “flow cell” possui na sua superfície dois tipos de oligonucleotídeos imobilizados complementares aos dois adaptadores, utilizados para produzir a biblioteca de sequenciamento. Estes oligonucleotídeos hibridizam com as moléculas das cadeias das bibliotecas. Por síntese reversa, começando pela zona hibridizada, a nova molécula que está sendo criada encontra-se covalentemente ligada à flow cell (Figura 3).
Esta nova molécula dobra-se e liga-se a outro oligonucleotídeo complementar ao segundo adaptador que não está ligado à placa, podendo ser usado para sintetizar uma segunda cadeia ligada também covalentemente à placa. Este processo de dobra da molécula e de síntese reversa, chamada de amplificação em ponte, é repetida várias vezes e cria aglomerados de milhares de cópias da sequência original, muito próximos na célula de fluxo.
Estes aglomerados distribuídos aleatoriamente contêm cópias idênticas da mesma sequência. Deste modo as bibliotecas estão prontas para serem sequenciadas.
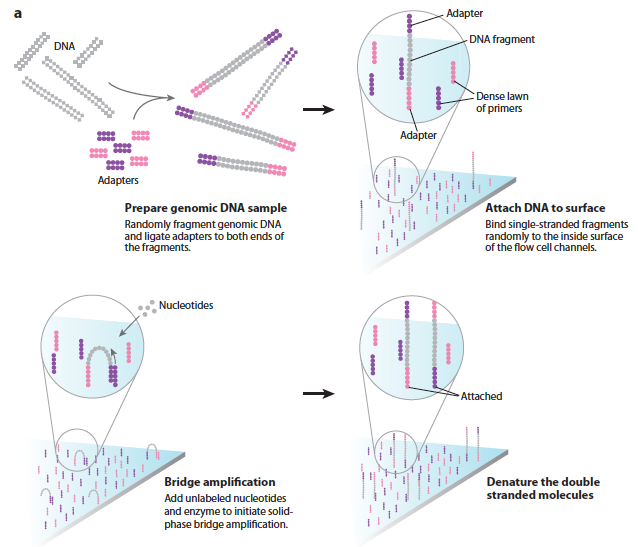
Figura 3: O processo de sequenciamento do Illumina.
No analisador de genoma, milhões de aglomerados são sequenciados simultaneamente. As moléculas de DNA são sequenciadas base a base em paralelo usando quatro nucleótideos marcados com fluorescência. As quatro bases completam umas com as outras para se ligar ao alvo, esta competição natural garante a alta precisão. Depois de cada síntese os fluorocromos são excitados por um laser, a cor obtida identifica a base que foi adicionada. Este fluorocromo é depois retirado para que a próxima base possa se ligar ao template, e então é lida cada base adicionada em cada ciclo.
REFERÊNCIA CONSULTADA
Carvalho, M. C. G. & Silva, D. C. G. 2010. Sequenciamento de DNA de nova geração e suas aplicações na genômica de plantas. Ciência Rural, vol. 40, Santa Maria.
Dick, G. J., Andersson, A. F., Baker, B. J., Simmons, S. L., Thomas, B. C., Yelton A. P., Banfield, J. F. 2009. Community-wide analysis of microbial genome sequence signatures. Genome Biology. v. 10, p. R85-1 a R85-16.
Duarte, E. A. Sequenciamento de segunda geração: Implicações nos estudos metagenômicos. Universidade Estadual de Santa Cruz.
Handelsman, J. 2004. Metagenomics: application of genomics to uncultured microorganisms. Microbiology and molecular biology reviews. v. 68, p.669-685.
Hoff, K. J., Lingner, T., Meinicke, P., Tech, M. 2009. Orphelia: predicting genes in metagenomic sequencing. Nucleic Acids Research. v. 37, p. W101-W105.
Kircher, M., Kelso, J. 2010. High-throughput DNA sequencing–concepts and limitations. Bioessays, 32, p.524-36.
Kunin, V., Copeland, A., Lapidus, A., Mavromatis, K., Hugenholtz, P. 2008. A bioinformatician’s guide to metagenomics. Microbiology and Molecular Biology Reviews. v. 557-578.
Mardis, E. R. 2008. Next-generation DNA sequencing methods. Annual Review of Genomics Human Genetics, 9, 387-402.
Moreno, A. C. A. 2013. Diagnóstico molecular na era da sequenciação de 3ª geração e da PCR digital. Dissertação de Mestrado. Universidade Fernando Pessoa.
Steele, H.; Streit, W.R. 2005. Metagenomics: Advances in ecology and Biotechnology. FEMS Microbiology Letters. v. 247, p. 105-111.
 Camarões Marinhos Reprodução, Maturação e Larvicultura (Livro Impresso)
R$50,00
Camarões Marinhos Reprodução, Maturação e Larvicultura (Livro Impresso)
R$50,00
 A produção integrada na carcinicultura brasileira (Volume 2) - (Livro Digital - PDF)
R$0,00
A produção integrada na carcinicultura brasileira (Volume 2) - (Livro Digital - PDF)
R$0,00
 As lendas na educação - Estórias do Baixo Sul e do Recôncavo Baiano (livro digital - PDF)
R$0,00
As lendas na educação - Estórias do Baixo Sul e do Recôncavo Baiano (livro digital - PDF)
R$0,00
 Um pouco de tudo o que você sempre quis saber sobre o cultivo de peixes orgânicos (livro digital - PDF)
R$0,00
Um pouco de tudo o que você sempre quis saber sobre o cultivo de peixes orgânicos (livro digital - PDF)
R$0,00
 Aquicultura no Brasil: o desafio é crescer (Livro Digital -PDF)
R$0,00
Aquicultura no Brasil: o desafio é crescer (Livro Digital -PDF)
R$0,00
 Água na indústria: uma análise da visão do setor industrial... (Versão Digital - PDF)
R$0,00
Água na indústria: uma análise da visão do setor industrial... (Versão Digital - PDF)
R$0,00
 Fichas Técnicas Ilustradas - Sebrae/Projeto AquiNordeste (Livro Digital - PDF)
R$0,00
Fichas Técnicas Ilustradas - Sebrae/Projeto AquiNordeste (Livro Digital - PDF)
R$0,00
 Piscicultura - Fundamentos e Técnicas de Manejo (Livro Digital - PDF)
R$0,00
Piscicultura - Fundamentos e Técnicas de Manejo (Livro Digital - PDF)
R$0,00
 As lendas na educação caiçara (livro digital - PDF)
R$0,00
As lendas na educação caiçara (livro digital - PDF)
R$0,00
 Atlas Anatômico e Histológico do Caranguejo-uçá (Livro Impresso)
O preço original era: R$70,00.R$55,00O preço atual é: R$55,00.
Atlas Anatômico e Histológico do Caranguejo-uçá (Livro Impresso)
O preço original era: R$70,00.R$55,00O preço atual é: R$55,00.
No dia 09 de fevereiro de 2021 foi finalmente assinado o Termo de Compartilhamento estabelecido entre a UFPR, através de...

O GIA acaba de lançar mais um livro. O tema da vez é o cultivo de peixes orgânicos Cada vez...