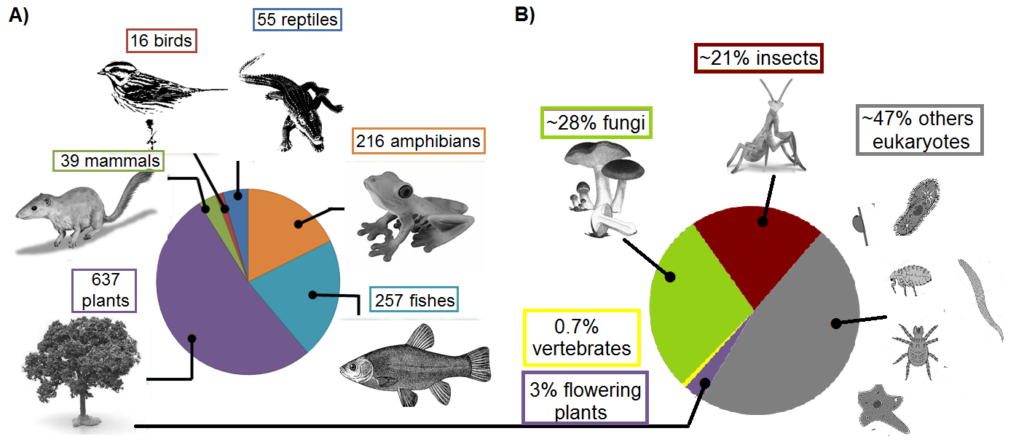
É importante destacar que, em geral, os dados moleculares derivados do “sequenciamento ambiental” devem ser vistos como complementares, ao invés de competir com os estudos taxonômicos. De fato, uma maior fusão de ambas as linhas de evidência é altamente justificada, pois será necessário superar suas respectivas deficiências (Dal Pont et al. 2021). Atualmente, partes da comunidade taxonômica dão pouca atenção (positiva) aos estudos de sequenciamento ambiental e vice-versa. É particularmente importante que os resultados da amostragem ambiental estejam disponíveis para os taxonomistas, pois podem incentivar o trabalho com linhagens não tratadas em uma estrutura taxonômica. Essa interação provavelmente criará “ciclos de retroalimentação taxonômica” que acelerará a descoberta de espécies e linhagens e potencialmente adicionará galhos proeminentes à árvore da vida. Assim, tal abordagem integrativa também melhorará a eficiência das estratégias de conservação, uma vez que mais esforços podem ser direcionados para “verdadeiros” hotspots abrangentes de biodiversidade, incluindo riqueza taxonômica e linhagens únicas em grupos “ocultos” combinando ambas abordagens taxonômica e moleculares de DNA ambiental.
Referencias:
Abarenkov, K. et al. 2016. Annotating public fungal ITS sequences from the built environment according to the MIxS-Built Environment standard – A report from a May 23-24, 2016 workshop (Gothenburg, Sweden). – MycoKeys in press.
Barrett, M. et al. 2018. Living planet report 2018: Aiming higher. in press.
Blaxter, M. et al. 2005. Defining operational taxonomic units using DNA barcode data.: 1935–1943.
Chapman, A. D. 2009. Numbers of living species in Australia and the world. in press.
Creer, S. et al. 2016. The ecologist’s field guide to sequence-based identification of biodiversity. – Methods Ecol. Evol. 7: 1008–1018.
Dal Pont, G. et al. 2021. Monitoring fish communities through environmental DNA metabarcoding in the fish pass system of the second largest hydropower plant in the world. – Sci. Rep. 11: 1–13.
Davies, N. et al. 2012. A call for an international network of genomic observatories (GOs). – Gigascience 1: 1–5.
Doliwa, A. et al. 2020. Identifying potential hosts of short-branch Microsporidia. – Microb. Ecol.: 1–5.
Elbrecht, V. et al. 2016. Testing the potential of a ribosomal 16S marker for DNA metabarcoding of insects. – PeerJ 4: e1966.
Hamilton, A. J. et al. 2010. Quantifying uncertainty in estimation of tropical arthropod species richness. – Am. Nat. 176: 90–95.
Maretti, C. C. 2014. Amazon: There is Hope! If we all do ‘the right thing’…; Deforestation, Protected Areas and Indigenous Territories: Past, evolution and… Which future?
Mora, C. et al. 2011. How many species are there on earth and in the ocean? – PLoS Biol. 9: 1–8.
Nilsson, R. H. et al. 2012. Five simple guidelines for establishing basic authenticity and reliability of newly generated fungal ITS sequences. – MycoKeys 4: 37–63.
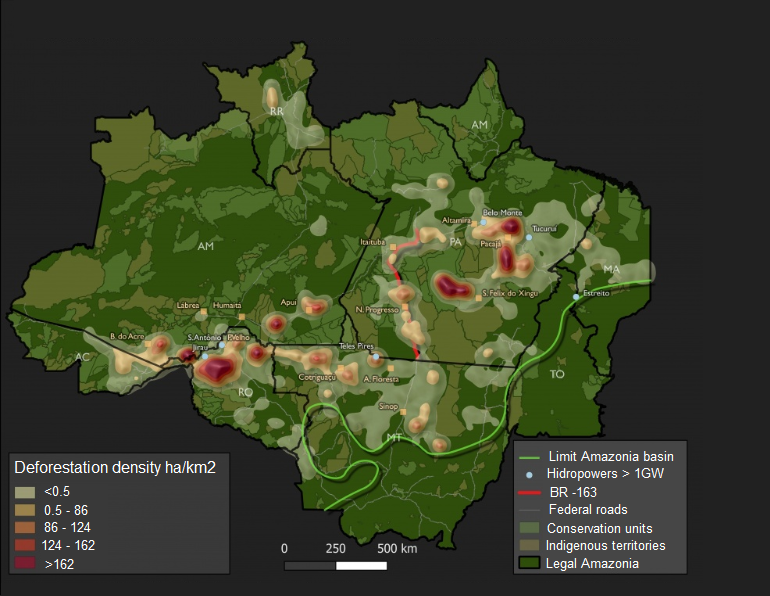
Pereira, E. J. de A. L. et al. 2019. Policy in Brazil (2016–2019) threaten conservation of the Amazon rainforest. – Environ. Sci. Policy 100: 8–12.
Prodan, A. et al. 2020. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. – PLoS One 15: e0227434.
Ritter, C. D. et al. 2019a. The pitfalls of biodiversity proxies: Differences in richness patterns of birds, trees and understudied diversity across Amazonia. – Sci. Rep. 9: 1–13.
Ritter, C. D. et al. 2019b. Locality or habitat? Exploring predictors of biodiversity in Amazonia. – Ecography (Cop.). 42: 321–333.
Ritter, C. D. et al. 2019c. Biodiversity assessments in the 21st century: The potential of insect traps to complement environmental samples for estimating eukaryotic and prokaryotic diversity using high-throughput DNA metabarcoding. – Genome in press.
Ritter, C. D. et al. 2020. Advancing biodiversity assessments with environmental DNA : Long-read technologies help reveal the drivers of Amazonian fungal diversity. 00: 1–16.
Tedersoo, L. et al. 2021. The Global Soil Mycobiome consortium dataset for boosting fungal diversity research. – Fungal Divers. 111: 573–588.
Ward, B. B. 2002. How many species of prokaryotes are there? – Proc. Natl. Acad. Sci. 99: 10234–10236.
Wearn, O. R. et al. 2012. Extinction debt and windows of conservation opportunity in the Brazilian Amazon. – Science (80-. ). 337: 228–232.
Yilmaz, P. et al. 2014. The SILVA and “all-species Living Tree Project (LTP)” taxonomic frameworks. – Nucleic Acids Res. 42: 643–648.

 Manual de ostreicultura com espécies nativas da região Nordeste do Brasil (livro digital - PDF)
R$0,00
Manual de ostreicultura com espécies nativas da região Nordeste do Brasil (livro digital - PDF)
R$0,00
 Rastreabilidade na ostreicultura: conceitos, fundamentos e casos... (livro digital - PDF)
R$0,00
Rastreabilidade na ostreicultura: conceitos, fundamentos e casos... (livro digital - PDF)
R$0,00
 As lendas na educação caiçara (livro digital - PDF)
R$0,00
As lendas na educação caiçara (livro digital - PDF)
R$0,00
 Aquicultura no Brasil: o desafio é crescer (Livro Digital -PDF)
R$0,00
Aquicultura no Brasil: o desafio é crescer (Livro Digital -PDF)
R$0,00
 A produção integrada na carcinicultura brasileira (Volume 1) - (Livro Digital - PDF)
R$0,00
A produção integrada na carcinicultura brasileira (Volume 1) - (Livro Digital - PDF)
R$0,00
 Camarões Marinhos Reprodução, Maturação e Larvicultura (Livro Impresso)
R$50,00
Camarões Marinhos Reprodução, Maturação e Larvicultura (Livro Impresso)
R$50,00
 Piscicultura - Fundamentos e Técnicas de Manejo (Livro Digital - PDF)
R$0,00
Piscicultura - Fundamentos e Técnicas de Manejo (Livro Digital - PDF)
R$0,00
 As lendas na educação - Estórias do Baixo Sul e do Recôncavo Baiano (livro digital - PDF)
R$0,00
As lendas na educação - Estórias do Baixo Sul e do Recôncavo Baiano (livro digital - PDF)
R$0,00
 Água no setor industrial da região do Alto Iguaçu e dos afluentes do Alto Ribeira... (Versão Digital - PDF)
R$0,00
Água no setor industrial da região do Alto Iguaçu e dos afluentes do Alto Ribeira... (Versão Digital - PDF)
R$0,00
 Manual de Boas Práticas: Qualidade e Segurança para Bons Negócios (livro digital - PDF)
R$0,00
Manual de Boas Práticas: Qualidade e Segurança para Bons Negócios (livro digital - PDF)
R$0,00
